






Techniques commonly used in various lab projects
- Genetically engineered mouse models (GEMMs).
- Retroviral transformation and transplantation of mouse hematopoietic stem cells using oncogenes found in leukemia patients
- Patient-derived xenografts (PDXs) by transplanting clinical samples from pediatric and adult leukemia patients into immunodeficient mice.
- Genome-wide genetic and epigenetic analysis (RNAseq, ChIPseq, ATACseq).
- Genome editing of established cell lines using CRISPR/Cas9.
- Molecular and cellular models to study gene expression and target validation.
Transcriptional Regulation of Hematopoietic Stem Cells and Immune Cells
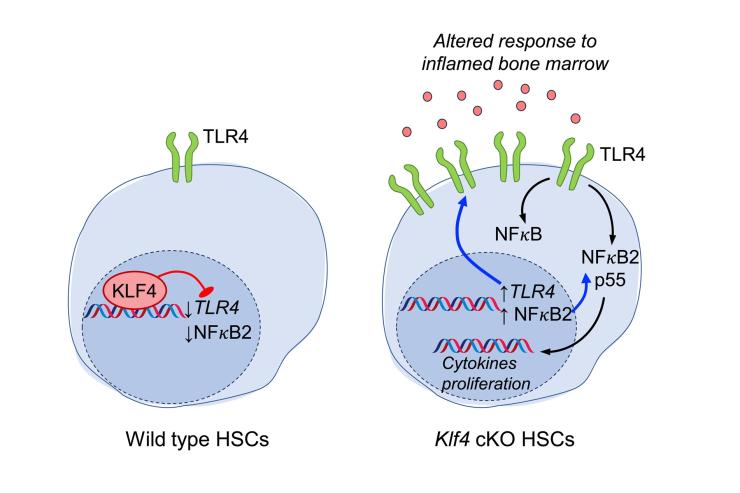
The Lacorazza lab studies how genetic networks regulate the behavior of hematopoietic stem cells (HSCs), a rare cell population that gives rise to the entire blood system. The study of HSCs is relevant to bone marrow transplantation, immune regeneration, and the development of leukemia. In a landmark study, we discovered that the transcription factor ELF4 maintains this delicate balance by releasing HSCs from quiescence (Cancer Cell, 2006). Building on this, we recently found that the Krüppel-like factor 4 (KLF4), a well-known reprogramming factor in the generation of induced pluripotent stem cells, preserves HSC regenerative potential after transplantation by suppressing toll-like receptor signaling and the non-canonical NFkB2 pathway during homeostasis (Experimental Hematology, 2025) (Figure 2). The regulatory power of ELF4 extends beyond stem cells. We showed that ELF4 restricts the proliferation of naïve T cells by upregulating KLF4 (Nature Immunology, 2009) and primes natural killer (NK) cells for rapid response (Immunity, 2002). In collaboration with Dr. Jordan Orange’s team, we identified hypomorphic ELF4 mutations in patients with NK cell deficiency, thereby directly linking our mouse model findings to human disease (JCI, 2022). We are currently investigating the role of inflammatory HSCs caused by KLF4 deficiency in the development of hematological pre-malignancies and immune response.
Genetic and Epigenetic Regulation in Lymphoid and Myeloid Leukemias
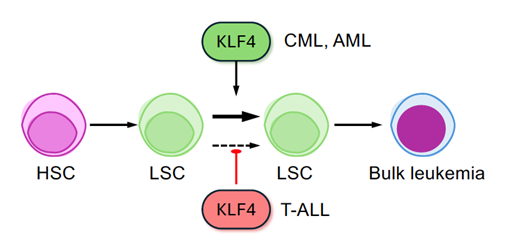
Can the same gene both guard against cancer and promote it? In our lab, we investigate this biological paradox through the study of KLF4, a transcription factor known for maintaining self-renewal in embryonic and cancer stem cells, in various types of blood cancer (Figure 3). In pediatric T-cell acute lymphoblastic leukemia (T-ALL), KLF4 acts as a tumor suppressor, limiting the expansion of aggressive leukemia-initiating cells (Leukemia, 2017). In contrast, in chronic myeloid leukemia (CML), the same factor functions as an oncogene, promoting leukemia stem cell self-renewal by repressing key inhibitory pathways (Blood, 2019). Similarly, KLF4 increases the frequency of leukemia-initiating cells in acute myeloid leukemia (AML) without affecting leukemia initiation (Stem Cells, 2022). By dissecting this duality, we aim to gain a deeper understanding of the context-dependent functions of KLF4 and identify new therapeutic strategies that selectively target leukemic stem cells. We are currently studying the role of KLF4 and other genetic and epigenetic drivers in leukemia using GEMM and pre-clinical PDX mouse models to translate our findings to human disease.
Targeting Relapsed Pediatric Leukemia
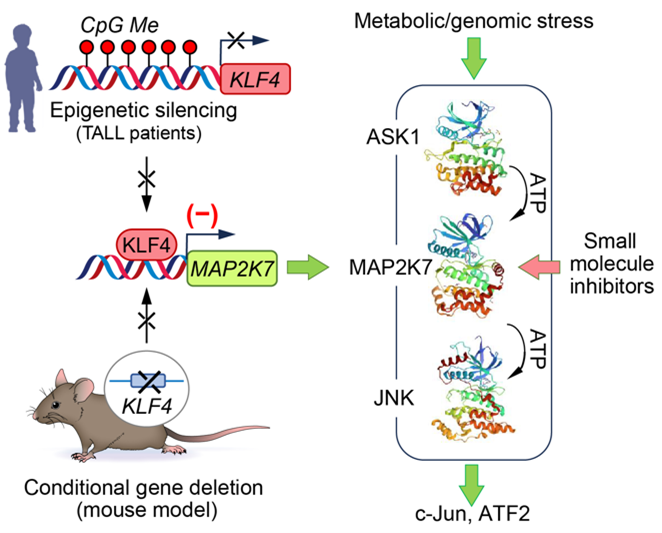
T-cell acute lymphoblastic leukemia (T-ALL) is one of the most aggressive blood cancers affecting children. While initial treatment is often effective, relapse or resistance to therapy dramatically worsens prognosis. Our lab is focused on understanding the molecular underpinnings of relapse disease to develop more effective and less toxic treatments. Using a genetically engineered mouse model of NOTCH1-driven T-ALL, we found that loss of the KLF4 gene results in a more aggressive disease phenotype. This is marked by the expansion of leukemia-initiating cells and activation of the stress-response kinase MAP2K7 (Leukemia, 2017). Strikingly, similar molecular alterations are observed in pediatric T-ALL patients, where KLF4 is silenced by DNA methylation, and MAP2K7 is pathologically activated (Figure 4). Building on these findings, we are now testing whether targeted inhibition of MAP2K7 can suppress leukemic growth without impairing normal T cell development (Blood Advances, 2023). This project is a collaborative effort with the Center for Drug Discovery at Baylor College of Medicine and medicinal chemists at Northwestern University, who are designing selective MAP2K7 inhibitors (ACS Med. Chem. Lett., 2023). We are also investigating the role of MAP2K7 and epigenetic landscapes in pediatric T-ALL and myeloid leukemias, as well as naïve T cells.
Targeting Leukemic Stem Cells in CML: A Path Toward Treatment-Free Remission
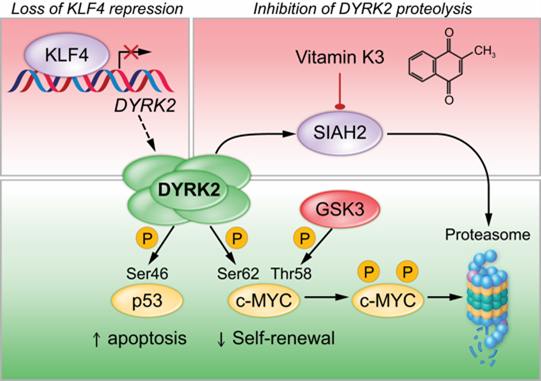
Our long-term goal is to develop a new class of precision therapies that eradicate LSCs, enabling durable, treatment-free remission for patients with CML. Chronic myeloid leukemia (CML) arises when hematopoietic stem cells (HSCs) acquire the BCR-ABL1 fusion gene, which encodes a constitutively active tyrosine kinase. Tyrosine kinase inhibitors (TKIs) have dramatically improved outcomes for CML patients, yet they fall short of a cure. Most patients must remain on therapy indefinitely, as leukemic stem cells (LSCs) evade treatment and can drive relapse if treatment is discontinued. Achieving treatment-free remission (TFR) requires novel strategies to eliminate these persistent, therapy-resistant cells. Our lab is addressing this challenge by identifying molecular vulnerabilities unique to CML LSCs. We found that the transcription factor KLF4 represses DYRK2, a dual-specificity kinase with tumor-suppressive activity in LSCs (Blood, 2019). DYRK2 is also targeted for degradation by the proteasome, limiting its therapeutic potential (Figure 5). By genetically or pharmacologically stabilizing DYRK2, we observed potent anti-leukemic effects in both genetically engineered mouse models (GEMMs) and patient-derived xenografts (PDX). To advance this strategy, we are developing cell-based drug screening platforms to identify compounds that stabilize the DYRK2 protein in LSCs, thereby establishing a therapeutic approach to eradicate chemoresistant LSCs.