






Preclinical Drug Development of mitochondrial-targeted therapies for TNBC
We are ‘translating’ our mechanistic understanding of mitochondrial rewiring and homeostasis in TNBC to devise therapeutic strategies to overcome chemoresistance. To achieve this, we have robust collaborations with multiple pharmaceutical companies to do therapeutic testing of novel agents, some which have already entered Phase I clinical trials, in our models of TNBC. We are also repurposing drugs that are already FDA approved for other applications. These studies combine mechanistic molecular biology with PDX preclinical trials and computational biomarker analyses. We collaborate with breast medical oncologists to plan clinical translation strategies. Figure made with Biorender.
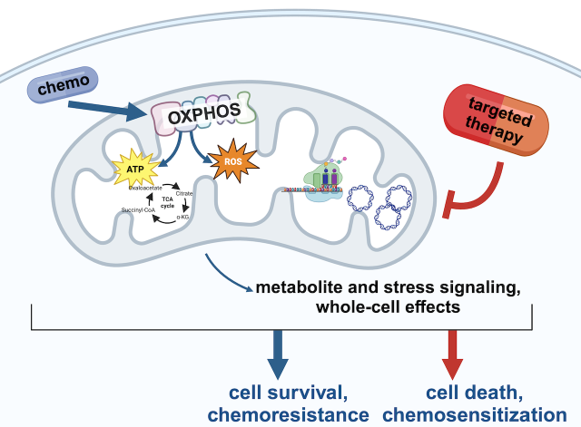
Mitochondrial Nucleic Acids: importance for TNBC chemoresistance and stress signaling
MtDNA and mtRNA transcribed from it primarily function to support cellular energy production through the synthesis of oxphos proteins within mitochondria. However, stress conditions such as chemotherapy can lead to the leakage of these nucleic acids into the cytosol. We are examining how this release occurs and how cytosolic sensors detect these nucleic acids, triggering survival mechanisms in chemotherapy-treated cancer cells. These pathways appear to mimic immune responses similar to those activated during viral infections, enabling chemoresistant cells to persist. Understanding the mechanisms behind stress-induced release and detection of mitochondrial nucleic acids may offer new strategies to overcome therapy resistance.
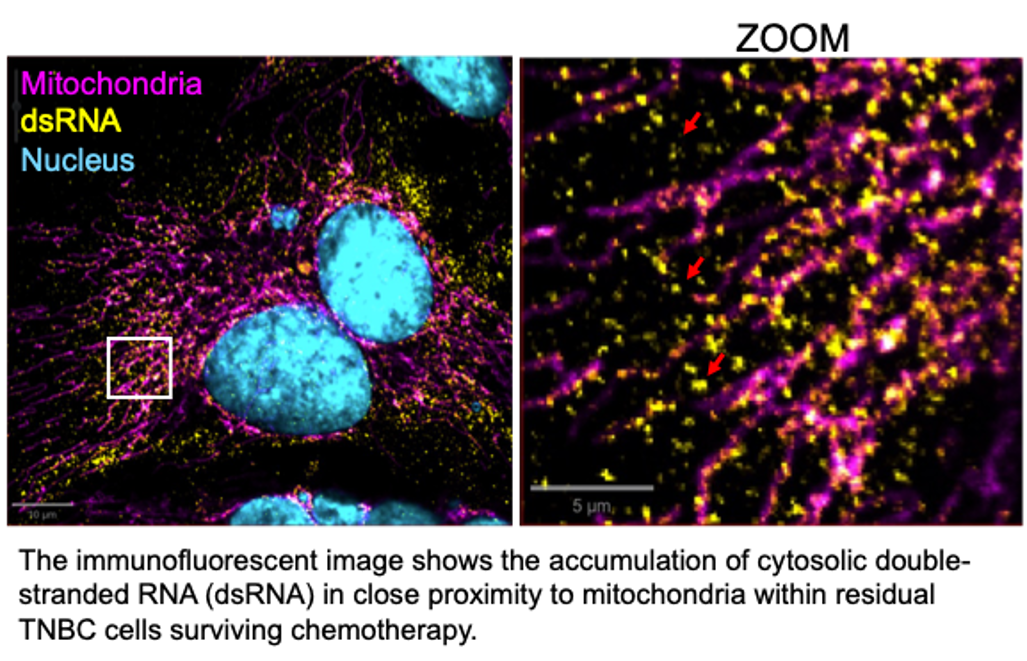
Lipid Storage and Acetyl-CoA axis supports TNBC chemoresistance
TNBC cells that survive chemotherapy dramatically increase levels of Acetyl-CoA, a key molecule for multiple metabolic, signaling, and epigenetic processes. This is accompanied by substantial accumulation of lipid droplets in the cytosol. We are exploring mechanisms driving lipid droplet formation and accumulation, and how we may perturb Acetyl-CoA metabolism to diminish lipid droplets and abrogate chemoresistance.

Mitochondrial structure: 3D analysis in TNBC therapeutic resistance
Mitochondrial structure dynamics regulated by mitochondrial fusion and fission, have been documented in various laboratory cancer models, yet there is limited understanding about the relevance of 3D mitochondrial structure in the context of therapeutic resistance. We are investigating the mitochondrial network dynamics between treatment-naïve and residual orthotopic PDX tumors persisting after exposure to conventional chemotherapies using state of the art 3D serial block facing scanning electron microscopy. This gives us unprecedented insights into the complexity and intra-tumoral heterogeneity of the mitochondrial networks in TNBC.
Protein Translation by the Mito-ribosome supports TNBC chemoresistance and can be therapeutically targeted
The mitochondrial respiratory chain (MRC) complexes are encoded by both nDNA and mtDNA. Mitochondria’s translation machinery is the mito-ribosome, which produces the mitochondrial proteins that contribute to the ETC complexes.
We find evidence for increased mitochondrial translation in chemoresistant cells surviving standard therapies. We are investigating the potential to therapeutically inhibit mitochondrial translation by repurposing conventional antibiotics whose mechanism of action is inhibition of the mito-ribosome.
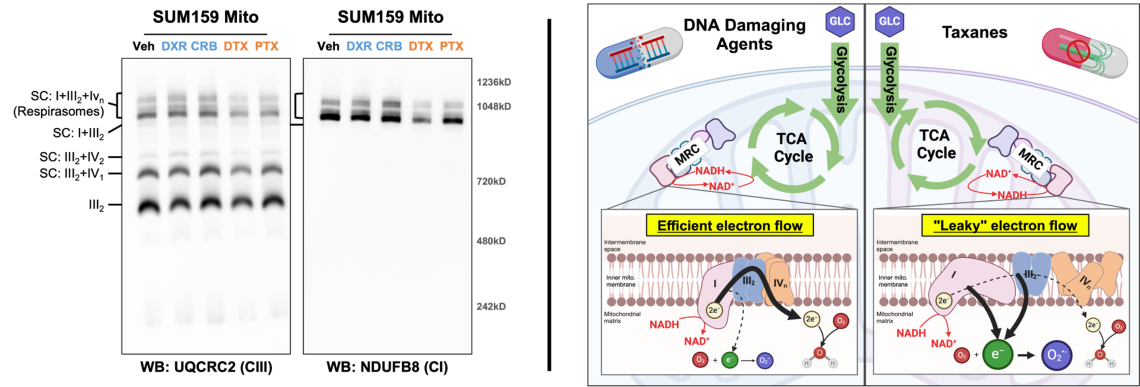
Chemotherapy class-specific alterations in mitochondrial respiratory chain formation may contribute to differential metabolic re
The mitochondrial respiratory chain (MRC) is comprised of four multi-subunit complexes which exist individually and aggregated with others known as supercomplexes (SCs). The formation and composition of SCs, particularly large SCs known as respirasomes, are thought to be an important regulator of electron flow through the MRC. TNBC cells surviving DNA damaging agents (e.g. doxorubicin and carboplatin) have elevated mitochondrial oxidative phosphorylation (OXPHOS) while cells surviving taxanes (i.e. docetaxel and paclitaxel) have diminished OXPHOS. Examination of MRC formation in residual TNBC cells showed respirasome formation is altered in a chemotherapy class dependent manner (left panel). These changes correlate with changes in OXPHOS and thus reveal a novel aspect of chemotherapy induced metabolic rewiring. We are diving deeper into the effects of each class of chemotherapy on the composition, stability, and function of the MRC.

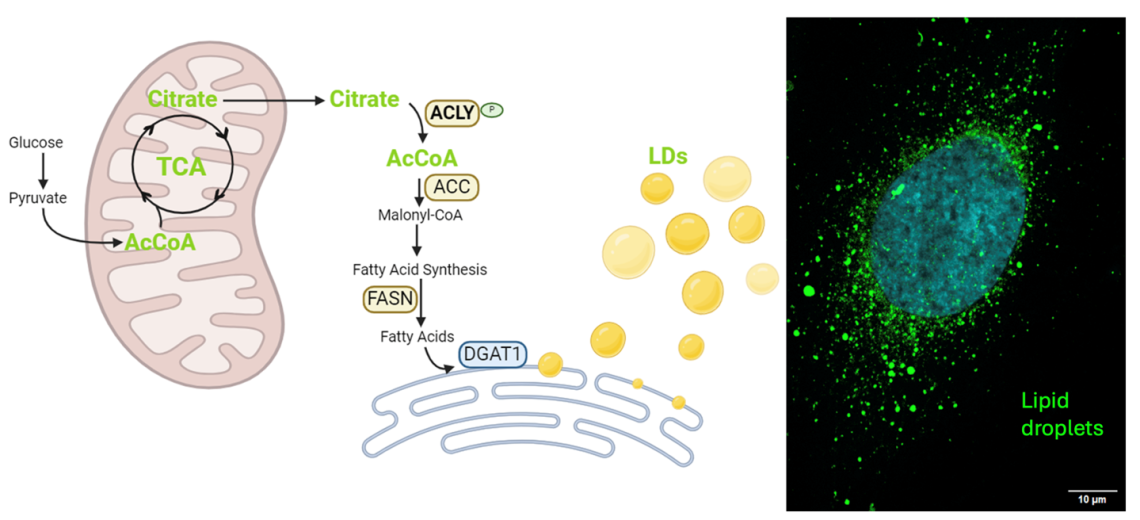
Post-transcriptional Regulation Via Rna Splicing And Proteolytic Cleavage of the mitochondrial fusion protein OPA1 in TNBC
We showed that mitochondrial fusion protein OPA1 drives mitochondrial fusion-promoted oxphos and chemotherapy resistance in TNBC. Regulation of OPA1 activity, while critically important for mitochondrial structure and oxphos, is poorly understood especially in cancer. OPA1 is regulated by alternative pre-mRNA splicing and proteolytic cleavage by mitochondrial proteases, producing 5 protein isoforms with distinct roles in the mitochondria. We are investigating how mitochondrial proteases and pre-mRNA splicing alterations are linked to TNBC mitochondrial function and therapeutic resistance via OPA1 using sophisticated genetic manipulations and protein analyses.
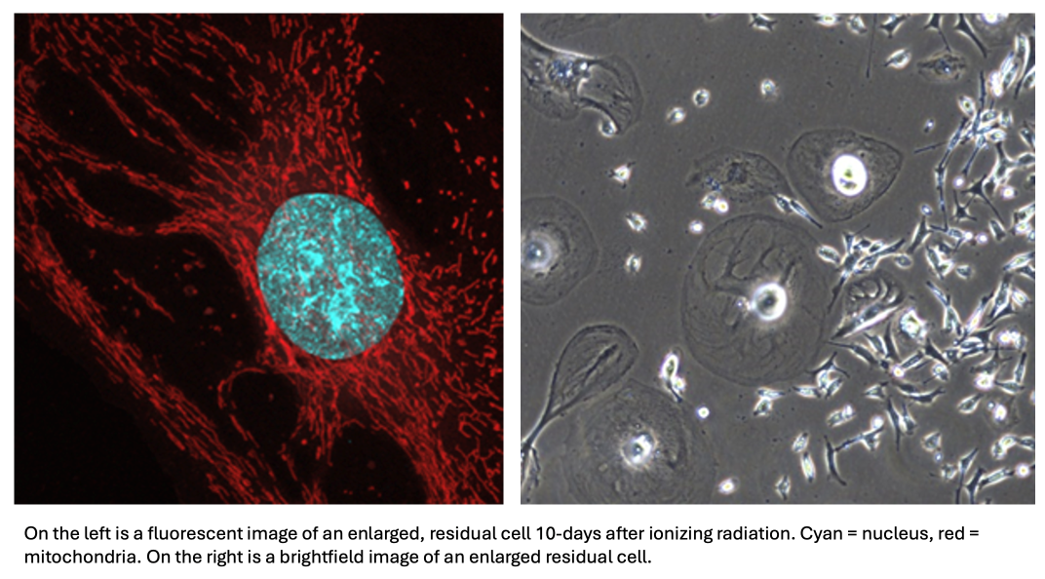
Radiation Therapy impacts mitochondrial and metabolic rewiring in TNBC
Our previous work with residual cells surviving DNA-damaging chemotherapies found that residual cells possess a host of mitochondrial adaptations, including increased oxidative phosphorylation, mitochondrial fusion, mitochondrial DNA copy number, as well as mitochondrial DNA damage. Given the mechanistic similarities between DNA-damaging chemotherapies and ionizing radiation, we aim to see whether our findings can be applied to the residual state after radiation, which is a common adjuvant therapy intervention for TNBC. Furthermore, we hope that investigating mitochondrial adaptations after a different DNA-damaging agent is applied will help us elucidate the potential pathways linking DNA-damage and these mitochondrial adaptations.

Spatial Intra-tumor Heterogeneity dynamics in TNBC chemoresistance
Utilizing spatial transcriptomic and metabolomic technologies, we are revealing distinct changes that occur in residual TNBC cells surviving chemotherapy in PDX models. The residual tumor has different proportions of transcriptomic clusters and metabolites across the tissue compared to the untreated and regrown. With these studies, we hope to reveal tumor-stroma cross talk mechanisms that may provide novel biological and therapeutic insights.